פרופ' ויויאן דרורי ודר' באטריס נפוסי, מרפאת ALS, מרכז רפואי תל-אביב
הידע על הבסיס הגנטי של ALS התרחב מאוד מאז שנת 1993, כאשר התגלה לראשונה שמוטציה בגן ,SOD1 שמקודד לאנזים סופראוקסיד דיסמוטאז, גורמת ל-ALS. האנזים SOD1 הוא חלבון שמפרק את המולקולות המזיקות של סופראוקסיד למי חמצן, שהם פחות מזיקים לתא. עד היום התגלו יותר מ-100 מוטציות שונות בגן זה. המוטציות אינן גורמות לשינוי בפעילות האנזים וכנראה שהן גורמות לפעילות מזיקה כלשהיא שעדיין, יותר מ-25 שנים לאחר זיהוי הגן, לא זוהתה. נמצא שהמוטציה גורמת לשינוי במבנה התלת מרחבי של החלבון ולכן נוצרים צברים של החלבון בתא, שאינם מפונים כראוי בשל פגיעה 'הקיימת בחולים אלו, במערכת האחראית לכך. מוטציות בגן זה מופיעות בכ-23% מהחולים עם ALS משפחתי (ידוע שבמשפחתם יש/היה חולה נוסף עם אותה מחלה), אבל הן נדירות מאד (כ-1%) בחולים ללא סיפור משפחתי.
בעקבות שכלול הטכנולוגיה של שיטות המחקר הגנטי, בעשור האחרון התגלו גנים רבים נוספים המעורבים במחלה וכיום מוכרים כ-70% מהגנים המעורבים ב-ALS משפחתי וניתן לזהות גנים לא תקינים אף ב-10- 15% מהחולים במחלה שאינה משפחתית (מחלה ספורדית). כמו כן, בחלק מהחולים נמצא יותר מגן פגום אחד. לכן, ככל הנראה, לפעמים נדרשים מספר גנים פגומים כדי לגרום למחלה.
התקדמות חשובה מאוד חלה בשנת 2011 עם הגילוי שמוטציה בגן הקרוי C9ORF72 גורמת ל-ALS. גן זה נמצא על כרומוזום 9 וכולל רצף קצר של נוקלאוטידים (אבני הבניה של הדנ"א) – GGGGCC – באנשים בריאים רצף זה חוזר על עצמו פעמיים עד 23 פעמים. באנשים עם המוטציה ב-C9ORF72, מקטע זה חוזר על עצמו מאות ואף אלפי פעמים (תמונה מס' 1). הגבול התחתון של מספר החזרות הגורם למחלה עדיין לא ידוע באופן ודאי, אך מקובל לחשוב שגן המכיל יותר מ-30 חזרות קשור למחלה.
בהמשך, מחקרים נוספים הראו שזוהי המוטציה השכיחה ביותר שגורמת ל-ALS באוכלוסייה המערבית. שכיחות של המוטציה נעה בין 23-47% בחולים עם ALS משפחתי, ובין 4-10% בחולים ללא עדות למחלה משפחתית. היא נמצאה בשכיחות הרבה יותר גבוהה באוכלוסיות חולים שמוצאם בצפון אירופה, לעומת דרום אירופה. והיא נדירה יותר בחולים ממוצא אסייתי או אפריקאי (לא יהודים).
מוטציה זו גורמת בשכיחות גבוהה גם לסוג מיוחד של שיטיון שקרוי Fronto-temporal dementia (FTD), המאופיין בשינויים באישיות, בהתנהגות ובתהליכי קבלת החלטות. שכיחות מוטציה זו נעה בין 20-35% בחולים עם FTD משפחתי ובין 4-8% בחולים ללא עדות למחלה במשפחה.
החשיבות הגדולה של גן זה, מלבד העובדה שזהו הגן השכיח ביותר שגורם ל-ALS והגן השכיח ביותר שגורם ל-FTD, הוא הגילוי המפתיע ששתי המחלות הללו, שיש להן תסמינים שונים לחלוטין, קשורות זו לזו. יש חולים שנושאים את המוטציה ויש להם שילוב תסמינים של ALS ושל ,FTD דבר המחזק את הטענה ש-ALS ו-FTD נמצאות על רצף קליני משותף. חיזוק נוסף לטענה זו נובע מממצאים פתולוגיים שמראים צברים של חלבון בשם TDP-43 בתאי העצב של רוב החולים שנושאים את המוטציה הזו, בין אם הם חולים ב-ALS או ב-FTD, או בשתי המחלות יחד.
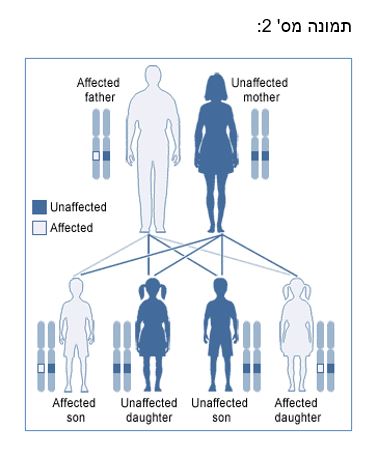
צורת ההורשה של המוטציה בגן זה היא אוטוזומלית דומיננטית, כלומר לאדם הנושא את הגן הפגום יש סיכוי של 50% להעביר את הגן הפגום לילדיו, כך שייתכן שאותו הילד יחלה, בשלב מסויים בחייו, אם כי אין זו חובה (תמונה מס' 2). חלק מהמחלות האוטוזומליות דומיננטיות בעלות חדירות לא מלאה ואז למרות נוכחות הגן הפגום, המחלה אינה מתבטאת. כאשר האדם נושא כרומוזום אחד עם המוטציה בגן C9ORF72, ההתבטאות הקלינית של המוטציה תלויית גיל ומוערכת באופן הבא: נדיר ביותר, קרוב לאפס אחוז עד גיל 35, 50% – בגיל 58, כמעט 100%- בגיל 80.
שכיחות התסמינים הראשונים בדיבור ו/או בבליעה בחוליALS שנושאים את המוטציה גבוהה יותר, בהשוואה לחולים שאינם נושאים את המוטציה. טווח גיל תחילת המחלה רחב ( 29-85 שנים) ותוחלת החיים שונה מאוד מאדם לאדם (3-96 חודשים מהסימנים הנוירולוגיים הראשונים). עדיין לא ברור האם יש קשר בין מספר החזרות של רצף הנוקלאוטידים לבין גיל תחילת המחלה, קצב התפתחותה או אופן התבטאותה. ככל הנראה קיימים גנים נוספים וגורמים סביבתיים שונים המשפיעים על גיל תחילת המחלה, צורת התבטאותה וחומרתה. רק עכשיו מתחילים לחקור באופן מעמיק יותר את הגורמים האלה.
כיום ניתן לבצע אבחון טרום לידתי ואפילו אבחון טרום השרשתי, אם ידוע על נשאות של המוטציה אצל אחד ההורים, או אחד מבני המשפחה. בכך ניתן למנוע את העברת הנטייה למחלה לדורות הבאים.
נחקרים שלושה מנגנונים אפשריים לנזק התאי שנגרם כתוצאה מהמוטציה: הפרעה בתהליך השעתוק של הדנ"א לרנ"א וכתוצאה מכך יצירת חלבון בלתי פעיל, אם כי עדיין לא ידוע מהו התפקיד של החלבון המקודד ע"י הגן C9ORF72; הרצפים החוזרים הרבים של דנ"א יוצרים רצפים רבים של רנ"א שמצטברים בגרעין תא העצב ונקשרים גם לחלבונים קושרי רנ"א ועלולים לגרום לנזק ; הרצפים החוזרים מתורגמים באופן בלתי תקין לחלבונים המכילים חזרות רבות של דיפפטידים (חלבונים קטנים) שמצטברים בציטופלסמה ועלולים גם הם לגרום לנזק. מחקרים עדכניים מראים שהנזק העיקרי לתאי העצב נגרם כנראה בגלל הצטברות של רנ"א ו/או חלבונים.
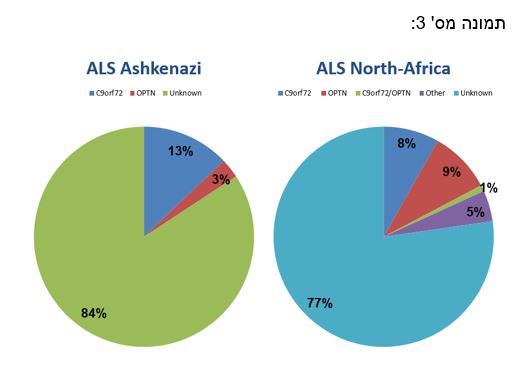
במסגרת פרויקט מחקר גדול על ממצאים גנטיים בחולי ALS בישראל המבוצע בימים אלה במרכז הרפואי תל-אביב ע"ש סוראסקי (איכילוב) בדקנו את שכיחות המוטציה בגן C9ORF72 בחולי ALS בשתי קבוצות אתניות של חולים יהודים בארץ – חולים ממוצא אשכנזי וחולים ממוצא צפון אפריקה. נמצא כי בחולים ממוצא אשכנזי %80 עם היסטוריה משפחתית ו-%11 מהחולים ללא היסטוריה משפחתית נשאו את המוטציה, בהשוואה ל- 0.3% בלבד באוכלוסייה הבריאה של יוצאי ארצות אשכנז. בקרב אוכלוסיית החולים ממוצא צפון אפריקה, 10% מהחולים עם היסטוריה משפחתית של ALS ו–9% מהחולים ללא היסטוריה משפחתית נשאו את המוטציה, בהשוואה ל-1% בקבוצת הביקורת הבריאה. (מאמר מס' 1)
בנוסף לשכיחות הגבוהה של הגן C9ORF72 בחולים בישראל, המחקרים שלנו הצליחו לגלות פגמים גנטיים נוספים, המסבירים יחד את סיבת המחלה ב-16% מהחולים האשכנזים ו-23% מהחולים יוצאי צפון אפריקה (תמונה מס' 3). לדוגמא, מוטציה בגן Optineurin נמצאה גם בחולים ממוצא אשכנזי וגם ממוצא צפון אפריקני, אמנם בשכיחות נמוכה יותר, אבל משמעותית. (מאמר מס' 2)
מוטציה נוספת, נדירה מאד בעולם, בגן המקודד לחלבון בשם Profilin 1 (PFN1) תוארה לראשונה במשפחה מישראל, יחד עם משפחה מאיטליה (מאמר מס' 3). גילוי פגם גנטי זה הביא להבנה שקיים מנגנון מחלה חשוב, שלא היה ידוע לפני כן – פגיעה ביציבות של שלד תאי העצב.
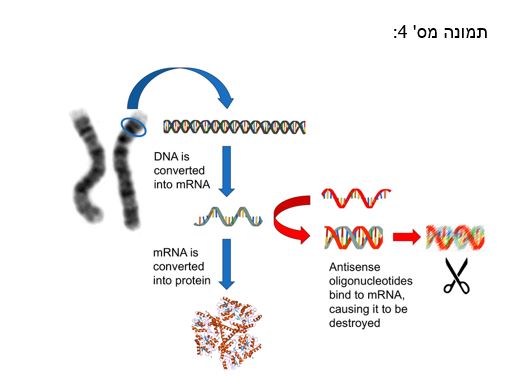
מספר חברות תרופות ומעבדות מחקר גדולות נמצאות כיום בתהליכי פיתוח מכוונים ל"השתקת" הגנים הפגומים ב-ALS SOD1) ו-C9ORF72), באמצעות טכנולוגיה חדשנית שמתבססת על מולקולות סינטטיות הקרויות אנטי-סנס אוליגונוקלאוטידים (antisense oligonucleotides – ASO). ASO הם מולקולות דנ"א או רנ"א, שרצף הנוקלאוטידים שלהם משלים לרצף הנוקלאוטידים של הרנ"א-שליח של הגן שאותו רוצים ל"השתיק"; הגדיל המשלים של ה-ASO נקשר לרנ"א שליח בתא ומונע את יצירת צברי רנ"א או חלבון. מולקולות ה-ASO יכולות לגרום להרס ספציפי של מולקולות הרנ"א שמשועתקות מהגן המוטנטי ובאופן זה למנוע היווצרות צברים טוקסיים של רנ"א, ו/או חלבונים שמקורם בגן המוטנטי (תמונה מס' 4).
הצלחה גדולה לטיפול התרופתי בטכנולוגיה זו הושגה עם מולקולות אנטיסנס לגן SMN1 שמוטציות בו גורמות לנוון שרירים שדרתי ( ,( Spinal Muscular Atrophy, SMAמחלה ניוונית תורשתית אחרת של תאי עצב מוטוריים. מחלה זו פוגעת בעיקר בתינוקות ובילדים עד גיל ההתבגרות וגורמת לחולשת שרירים קשה. כולל לפעמים חולשת שרירי הנשימה, נכות וקיצור ניכר של תוחלת החיים. עד כה לא היה טיפול למחלה גנטית זו, אך לפני שנתיים אושרה לשימוש תרופה ראשונה לטיפול במחלה, העובדת במנגנון של ASO. הילדים המטופלים לא רק נותרו בחיים, אלא שהיכולות המוטוריות שלהם השתפרו. כך שלראשונה יש טיפול שיכול להציל חיים ולשנות את מהלך המחלה, במחלה גנטית קשה של תאי עצב מוטוריים.
הסנונית הראשונה בפיתוח תרופות מבוססות טכנולוגיית אנטיסנס ב-ALS היא תרופה בשם Tofersen, שהיא תרופת אנטיסנס שמתמקדת בגן SOD1. תוצאות מחקר קליני ראשון, פאזה 1-2, כפול סמיות ומבוקר פלצבו, ב-48 חולי ALS שנושאים את הפגם הגנטי בגןSOD1 פורסמו לאחרונה (מאמר מס' 4). המחקר נערך במספר מרכזים רפואיים בארה"ב, קנדה, ומספר מדינות באירופה, וארך 12 שבועות של מתן טיפול ועוד 12 שבועות של מעקב. מטרתו העיקרית של המחקר הייתה לבדוק את השפעת הטיפול על ריכוז החלבון SOD1 בנוזל השדרה ואת בטיחות הטיפול, שניתן בהזרקה לתוך נוזל השדרה (5 הזרקות במהלך 12 שבועות). נבדקו 4 ריכוזים של התרופה ונמצא כי ריכוז החלבון SOD1 ירד בנוזל השדרה של המטופלים, כאשר הירידה הגדולה ביותר הייתה בחולים שקיבלו את המינון הגבוה ביותר של התרופה ועמדה על כ-36%, בהשוואה לחולים שקיבלו חומר דמה (פלצבו) (תמונה מס' 5). המחקר לא תוכנן כדי לבדוק את ההשפעה הקלינית של התרופה, היות והטיפול היה לתקופה קצרה מדי. ובכל זאת נראו עדויות לכך שבמינון הגבוה ביותר התרופה האטה את התקדמות המחלה, כפי שנבדק ע"י הציון במדד התפקודי ALSFRS-R, בתפקוד הנשימה ובמבחן כוח (תמונה מס' 6). בימים אלה נערך מחקר קליני פאזה 3, במספר גדול יותר של חולים, שיטופלו במשך שנה לפחות, שמטרתו היא לבדוק את יעילות הטיפול בהאטת המחלה. מחקר זה אמור להסתיים ביולי 2021.
בנוגע למוטציה בגן C9ORF72 – מחקרים מצאו שטכנולוגיית ASO מורידה את כמות צברי הרנ"א שמקורם במוטציה. בתרביות תאים ובמודלים של מכרסמים נמצא ש-ASO הורסים צברי רנ"א וצברי חלבונים בתאי עצב תנועתיים ויכולים אף למנוע היווצרות של צברי חלבונים חדשים. תרופת אנטיסנס למוטציה בגן C9ORF72 נמצאת כעת בשלב הניסויים הקליניים המוקדמים בבני אדם.
אף על פי שאין עדיין טיפול יעיל למחלה לכל החולים, ההבנות החדשות שנלמדו מהמחקרים הגנטיים של העשור האחרון והתוצאות המעודדות בשימוש בטכנולוגיות החדשניות נותנות תקווה שטיפול יעיל למחלה יפותח בעתיד הנראה לעין לרוב החולים. הטיפול יהיה מותאם באופן אישי לכל חולה וחולה על פי הרקע הגנטי של מחלתו, ולכן יהיה הרבה יותר יעיל מהטיפולים הקיימים היום.

מאמר 1:
Goldstein O, Gana-Weisz M, Nefussy B, Vainer B, Nayshool O, Bar-Shira A, et al. High frequency of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis patients from two founder populations sharing the same risk haplotype. Neurobiol Aging 2018; 64:160.
מאמר 2:
Goldstein O, Nayshool O, Nefussy B, Traynor BJ, Renton AE, Gana-Weisz M, et al. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology 2016;86:446-53.
מאמר 3:
Wu CH, Fallini C, Ticozzi N, Keagle P, Sapp PC, Piotrowska K, et al. Mutations in the Profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature, 2012; 488 (7412): 499-503.
מאמר 4:
Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC et al. Phase 1-2 trial of antisense oligonucleotide tofersen for SOD1 ALS. New Engl J Med 2020; 383:109-119.